

Venom from a lethal South American snake enabled the development of the first Angiotensin-converting-enzyme (ACE) inhibitor drugs – which have saved or improved the lives of millions of people with cardiovascular disease, high blood pressure and kidney disease. Although the first ACE inhibitor, captopril, is now rarely used, its multi-billion dollar successor lisinopril topped the US drug charts and is still the country’s fourth-most prescribed medication. In this article – the latest in our Pharmaceutical Roots series – we discuss ACE inhibitors’ history, uses, risks, and mechanisms of action, as well as the future therapeutic potential of medicines derived from snakes.
Introduction
Cardiovascular disease (CVD) is the leading cause of death globally, causing just under a third of all mortality in 2019, when it ended the lives of almost 18 million people. An umbrella term for conditions that affect the heart and circulation, CVD is most commonly associated with a waxy buildup that narrows or blocks blood vessels – leading to angina, high blood pressure, heart attacks, and strokes. Aside from ACEs, treatments for CVD include statins, beta blockers, angiotensin receptor blockers (ACBs) and lifestyle changes like regular exercise and stopping smoking. Surgical interventions – such as coronary angioplasty, stents, bypass surgery, and even heart transplants – may be considered in particularly serious cases.
In simple terms, ACE inhibitors treat hypertension and protect the heart by blocking the body from producing the hormone angiotensin II, which in turn reduces blood pressure by (a) causing blood vessels to dilate and (b) reducing the amount of water that the kidneys put back into the blood. However, the journey from snake venom to producing these life-changing drugs was anything but easy – and has been described as “one of the great success stories of modern medicinal chemistry.”
Fangs to pharmaceuticals
The Brazilian pit viper, also known as the yarará, or Bothrops jararaca, is a deadly, greyish-brown snake that grows up to 160cm long and can be found mainly in southern Brazil, northern Argentina, and northeastern Paraguay. Its venom brings about a drop in blood pressure that causes prey to lose consciousness, as well as other potentially fatal effects such as haemorrhage and incoagulable blood. In the early 1980s, speakers at hypertension conferences often livened up their presentations by replacing graphs and tables with pictures of the dangerous snake’s “striking zig-zag markings and aggressively protruding tongue”. But the effects of yarará venom had been safely harnessed for the good of medical research for around 15 years by then – notably as a peptide extract brought to Europe by the Brazilian researcher Sergio Ferreira. When tested in the laboratory of future Nobel Prize winner John R. Vane, Ferreira’s venom peptide “proved to be an ACE inhibitor and, while not suitable to be a drug, lowered blood pressure in clinical trials.” As well as kindling Vane’s “strong interest” in inhibiting ACE to treat hypertension, the extract also “provided an important clue to the structure of ACE inhibitors.”
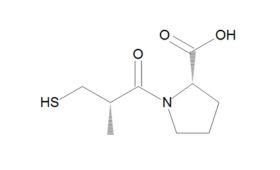
ACE had first been identified as responsible for converting angiotensin I to the vasoconstrictor angiotensin II in the mid-1950s. But producing a suitable synthetic and orally active analogue of Ferreira’s 1960s venom peptide would require another decade of “full-press, top priority” and expensive work by the US drug firm Squibb. From 1970 to 1973, Squibb scientists unsuccessfully tested around 2,000 chemical structures for ACE inhibitor activity – finally achieving a breakthrough in early 1974 by following up new research on an inhibitor of carboxypeptidase A, an exopeptidase thought to have a similar active site to ACE. After studying a further 60 compounds over the following 18 months, they finally synthesised captopril – which soon demonstrated its antihypertensive effects in clinical studies and was granted US Food and Drug Administration (FDA) approval in 1980. The new compound was a notable success: not only becoming Squibb’s first billion dollar drug, but also creating a foundation for the even more effective ACEs that came after it. As Philip Poole-Wilson, emeritus professor of cardiology at Imperial College, London, remarked: “The discovery of the ACE inhibitors and the creation of captopril was one of the really great advances in cardiovascular medicine.”
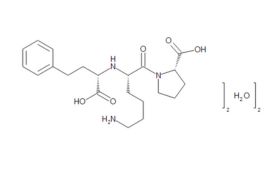
From captopril to lisinopril
Although a game-changer for treating CVD, captopril did not make optimum use of all available binding sites – prompting researchers to perform a series of structure-activity studies in the hope of finding even more effective treatments. These led eventually to FDA approval of the Merck company’s own ACE inhibitor, enalapril, in 1985. Scientists at Merck then systematically altered each structural unit of enalaprilat (the active metabolite of the enalapril prodrug), by substituting various amino acids, including lysine. Adding lysine not only gave the new compound twofold better binding ability than enalaprilat, but also provided a name for what would become the most successful ACE inhibitor drug of all time. First approved by the FDA in 1987, lisinopril went on to become America's most popular medicine, and was still fourth on the bestseller lists in 2021, with more than 88 million prescriptions in the US alone.
Captopril and lisinopril: mechanisms of action, dosages and differences
Captopril and lisinopril both work by suppressing the body’s renin-angiotensin-aldosterone system (RAAS) – lowering blood pressure by inhibiting ACE from converting angiotensin I to vasoconstricting angiotensin II, and also by decreasing aldosterone secretion that would otherwise cause the kidneys to reabsorb water and sodium. Furthermore, both drugs impede the degradation of bradykinin, a peptide that causes vasodilation, thus enhancing its blood pressure lowering effect.
Lisinopril is a long-acting drug administered once daily by mouth and, unlike captopril or enalapril, is hydrophilic and not broken down by the liver. Captopril is normally taken three times per day, and has a half-life of two hours. Side-effects of captopril include angioedema (swelling of the face, lips, arms, legs, tongue and throat that can obstruct breathing and potentially prove fatal), although lisinopril is well tolerated, with “a profile of adverse effects... typical of ACE inhibitors as a class.” However, both captopril and lisinopril may raise levels of potassium in the blood, and so care should be taken when prescribing them along with other treatments that increase potassium concentrations – such as diuretics and nifedipine.
Chemistry and synthesis

Snake venom based drugs: a fang-tastic future?
Many scientists believe that there is now a high probability of animal toxins like snake venom being exploited as drugs of the future. As a recent journal review puts it, venoms contain many biomolecules and inorganic compounds whose physiological and pharmacological activities could – like yarará venom - be used in ways that are unrelated to primary functions like prey immobilisation and defence. Moreover, venom toxins can act on a wide range of molecular targets – including membranes, proteins, receptors, and ion channels and are “increasingly getting exploited as pharmacological tools and prototypes for drug development because of their high degree of specificity.” Already, snake venoms have been identified as potential prototypes for new cancer drugs, as well as neurodegenerative-, autoimmune-, and infectious-parasitic diseases. As part of this trend, the yarará venom was re-investigated in a functional genomics/connectivity mapping study that indicated it may have potential for treating neuropsychiatric illnesses like Parkinson’s disease, schizophrenia, depression, and epilepsy, as well as possible anti-inflammatory and antimicrobial properties. Although many animal toxins offer ‘unquestionable’ biotechnological potential, snake venoms are perhaps the most promising of all – due to the variety of bioactive components they contain, but also because their greater availability compared to scorpions, spiders, wasps, and bees.
|
Author Matt Gregory – Global Product Manager
Matt holds a MSci in Chemistry from the University of Birmingham, as part of which he worked in early drug discovery, before stepping into process development and kilo scale API manufacture in a Current Good Manufacturing Process (cGMP) environment. He has worked at LGC Standards for two years, enabling him to further explore some of his significant interests in the pharmaceutical industry - from early stage hit- and lead generation to method development and validation.
|


{{ errors[0] }}